Information Technology Reference
In-Depth Information
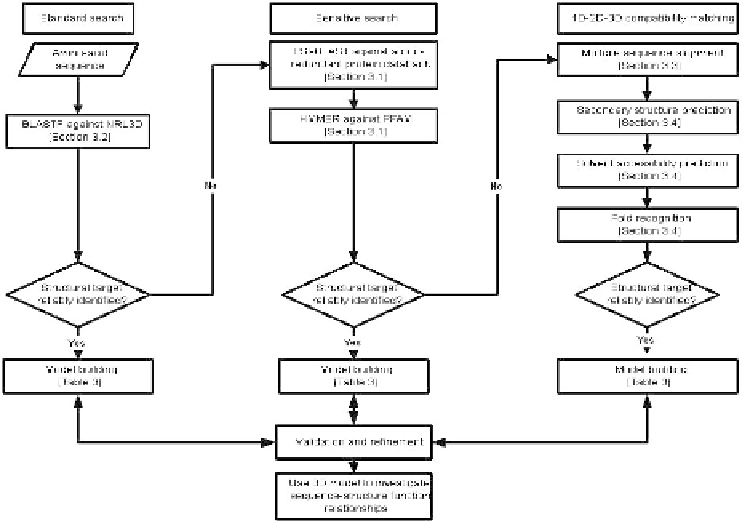
Figure 1.
A flowchart for predicting 3D structures from protein sequences by using
bioinformatics techniques. Predictions using “standard searches” are the most
accurate. The sensitive searches and “1D-2D-3D” compatibility matching methods
are non-trivial methodologies that can sometimes add value to the sequences where
the standard techniques do not identify a structural template for the query sequence.
The first step in a typical protein structure prediction is to establish if a protein
sequence or part of a protein sequence has any homologues of known structure in the
Protein Data Bank (PDB) [17, 18]. Typically, protein structures are experimentally
determined and classified at the level of the domain [19, 20]. Comparative molecular
modelling or homology modelling is currently the most successful and accurate method for
protein structure prediction [21]. If a protein structure prediction can be based on comparative
molecular modelling (Table 3), this should be the method of choice (Figure 1). In the absence
of high sequence identity between sequence and structural homologues, deciding what
constitutes significant sequence similarity is not straightforward. This type of prediction
then becomes “non-trivial”. The most promising methods for solving this type of problem
involves performing sensitive sequence searches and characterising sequence compatibility
with the structural properties of known secondary and tertiary protein structure (also known
as “1D-2D-3D” compatibility matching methods). Sensitive searches help identify weak
similarities between the sequence of interest and homologues that have had their structures
experimentally determined to atomic resolution. The “1D-2D-3D” compatibility matching
methods include, secondary structure and solvent accessibility predictions as well as protein
fold recognition. Such methods can be useful in predicting common structural folds for
proteins that share little or no sequence similarity (Figure 1). However, at low levels of
sequence similarity the structures of proteins sharing a common fold diverge to such an
extent that the accuracy of models built by comparative techniques are significantly
reduced [21, 22].