Biology Reference
In-Depth Information
Genome annotation
(new gene models derived from proteomics data)
exploratory
Proteomics
targeted
Proteomics
ProMEX
(protein-/proteotypic peptide database)
MAPA
nano UPLC-Orbitrap-MS
(extraction of accurate precursor ions m/z, spectral
count or intensity, samples versus variables
alignment)
MASS WESTERN
nano UPLC-TripleQuadrupole-MS
(synthetic stable isotope-labeled internal
standards, absolute protein concentrations)
COVAIN
Data integration/PCA/ICA/Clustering/Granger
causality/Correlation networks/differential
Jacobian
biomarker/physiological marker/correlation networks/
sample pattern recognition/
statistics/modeling/metabolic regulation
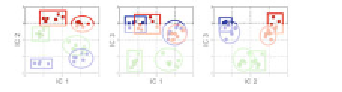
Fig.
1
Overview of a proteomic-toolbox for systems biology. A central proteome/peptide spectral database
(ProMEX,
www.promexdb.org
[
8
,
9
]) serves as a basis for the selection of proteotypic peptides, suitable for the
targeted MASS WESTERN analysis [
4
] of complex proteome samples (
see
Fig.
3
). The MAPA method
(MAPA = Mass Accuracy Precursor Alignment [
3
]) allows for the detection and quantifi cation of all changes of
tryptic peptides
m
/
z
-ratios in the LC-MS shotgun analyses independent of database search. Quantitative pro-
teomics data are aligned by the ProtMAX algorithm and statistically analyzed using the COVAIN toolbox [
5
].
Proteomic data enable the suggestion of new gene models not found via computational prediction from the
raw genome sequence alone [
20
]. All these data as well as metadata (experiment, parameters of analysis, and
database search) are stored in the database ProMEX. Furthermore, raw spectra from MAPA analysis can be
searched against PROMEX [
8
,
9
]
also in human proteomics following numbers are being antici-
pated: Assuming approximately 20,000-30,000 annotated genes
of a genome, after consideration of splice-variances and posttrans-
lational modifi cations such as phosphorylation or glycosylation,
several hundreds or thousands of possible protein species per anno-
tated gene may be reached. It is, however, not assumed that all
possible protein isoforms are active at the same time. Nevertheless,
existing technologies are confronted with enormous challenges
due to high number and dynamic concentration range of all pro-
teins of a single steady state. For gaining a holistic overview of the
dynamics of a continuously transient biological system it is impor-
tant to analyze many different steady states, time series, diverse
genotypes, and their phenotypic plasticity. In modern biology, a