Biomedical Engineering Reference
In-Depth Information
+
Catalase (CAT)
H
2
O
O
2
Glutathione reductase (GSR)
Pathologic
Loading
Mitochondrial
Electron Transport
GSSG
GSH
H
2
O
2
H
2
O
O
2
Glutathione peroxidase
(GPX)
Superoxide
Dismutase
(SOD)
Inflammation
NADPH Oxidase
Peroxiredoxin (PRDX)
Txn
-S
2
Txn
-(SH)
2
Thioredoxin reductase
Fig. 4 Theoretical mediators of obesity-dependent ROS production. The effect of obesity per se
on chondrocyte ROS production is not well known. However, both pathologic (traumatic)
mechanical loads and inflammatory mediators are known to increase the generation of superoxide
anion (O
2
-
). Impaired mitochondrial function and activation of NAPDH oxidase enzymes are
two primary sources of ROS production in chondrocytes. Superoxide anion is metabolized by
superoxide dismutase into hydrogen peroxide (H
2
O
2
), which is further metabolized into water
and oxygen through one of several antioxidant enzymatic pathways (antioxidants shown in
green). It is important to note that these reactions occur in sub-cellular compartments and are thus
dependent
on
the
activity
of compartment-specific
antioxidant
isoforms.
A
more
detailed
discussion of chondrocyte ROS production and metabolism can be found in [
106
]
production and oxidative damage were derived from diseased waste tissue
obtained from obese patients. However, whether or not ROS-dependent disease
mechanisms vary with obesity status and contribute to the increased risk of OA
remains to be determined. Two likely mechanisms by which obesity increases
ROS production are altered mitochondrial function and NADPH oxidase activa-
tion. In both cases, biomechanical stimulation and inflammation are likely con-
tributing factors to altered ROS production (Fig.
4
).
Mitochondrial ROS generation is a normal process of ATP production in the
mitochondria due to electrons escaping from the electron transport chain and
interacting with molecular oxygen to form superoxide anion. Although oxidative
phosphorylation is a relatively minor source of ATP production in chondrocytes
(\&25 % of total ATP production), preventing the entry of electrons into the
electron transport chain at complex I reduces chondrocyte ROS production as
measured by oxidation of dihydroethidium [
110
]. This basal level of mitochondrial
ROS generation appears to play an important role in maintaining cellular energetic
homeostasis by stimulating and/or stabilizing non-oxidative glycolysis [
110
].
Physiologic ROS also appears to be important in facilitating ion homeostasis in
chondrocytes [
111
]. During physiologic levels of compressive loading, mitochon-
drial ROS production is increased [
112
]. Reducing or scavenging mitochondrial
ROS production during loading using Rotenone and MitoQ, respectively, reduces
cellular oxidation and ATP levels [
112
]. In contrast, traumatic levels of compressive
impact loading also increase mitochondrial ROS generation, but under these con-
ditions, excess mitochondrial ROS increases cell death [
113
]. These findings sug-
gest that a continuum exists whereby physiologic compressive loading stimulates






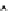



























Search WWH ::

Custom Search