Biology Reference
In-Depth Information
well as issues encountered applying the approach
for peptide analysis will also be discussed.
isotope-labeled internal peptide standard spiked
in with known amount.
The targetedMS-based approach to be applied
to biomarker veri
cation
of peptides derived from the protein biomarker
candidates. Once the protein biomarker can-
didates to be veri
cation relies on quanti
MRM-MS ASSAY GENERATION FOR
PROTEIN QUANTITATION
ed are determined, the
peptides targeted for their accurate quantitation
have to be unique in the proteome to these
speci
Multiple-reaction monitoring mass spectrom-
etry (MRM-MS) is generally done using triple
quadrupole or hybrid triple quadrupole ion
trap mass spectrometers because of their short
duty cycle as well as their linear quantitation
range. MRM-MS is performed by isolating the
precursor ion in the
c proteins. Peptides and subsequent transi-
tions suitable for MRM-MS assays must be evalu-
ated and optimized to achieve best performance
with least interferences. In fact, peptides seen in
discovery stages are not necessarily the best can-
didates to give best performance in MRM-MS
assays. Additional optimization of MRM-MS
experiments are still required to narrow down
the best peptides and/or the best transitions to
monitor in the interested sample matrix to
achieve the best sensitivity and reproducibility.
The best way to generate optimized MRM-MS
assays obviously is to start with recombinant
proteins if they are available. However, in most
cases, the recombinant proteins are not available
and/or too expensive/dif
first quadrupole (Q1), frag-
menting it within Q2, and monitoring the
optimum fragment ions using Q3. The selectivity
and speci
city is achieved through a combina-
tion of Q1 isolating only precursor peptide ions
within a narrow mass window and monitoring
fragment
ion masses corresponding to the
speci
c precursor ions in Q3. The selectivity typi-
cally increases when multiple transitions are
monitored for the same precursor ion, with
three to
five transitions typically being used
for selectivity purpose. As shown in
Figure 1
,
absolute quantitation is achieved using a combi-
nation of stable isotope dilution (SID) and
MRM-MS by comparing the peak area obtained
from the endogenous peptide (best performing
transition peak area or peak area from combined
transitions) and that obtained from the stable
cult to generate. Pep-
tides observed during discovery stage and/or
selected from predicated high-responding
peptides after an in silico proteolytic digestion
13
after further
filtering using certain criteria are
used to generate the MRM-MS assay. Besides
using recombinant proteins, crude synthetic
Protein
Q3
Q1
Q2
Area A
k
k
k
LC
digest
k
k
k
Area B
k
k
k
k
Precursor isolation
CID
Fragment(s) isolation
Heavy stable isotope standard
FIGURE 1
MRM-MS method development and optimization workflow for protein quantitation.









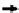



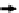

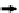




Search WWH ::

Custom Search