Biology Reference
In-Depth Information
A: Feature detection
de-isotoping
3D peak detection
12
C
retention time
alignment
13
C
13
C
2
retention
time
mass
recalibration
m/z
m/z
B: Peptide identification
C: Protein identification
b
1
b
2
b
3
b
4
b
5
b
6
PEP T I D E
y
6
y
5
y
4
y
3
y
2
y
1
non-unique
peptide
sequence
database
search
unique
peptides
score calculation
target-decoy-based
FDR thresholding
peptide to protein mapping
FDR thresholding
D: Quantification
label-free
stable isotope-based
SILAC, dimethyl
compare intensities across runs
Δm/z
retention
time
reporter ion-based
iTRAQ, TMT
MS/MS
run #2
m/z
run #1
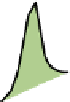
FIGURE 1.2
Overview of the main components of the computational workflow of shotgun proteomics. A: Detection and processing of peptide
features in LC-MS runs. B: Identification of peptides based on their characteristic fragmentation patterns. C: Assembly of peptides into proteins. D:
Quantification of peptides and proteins based on stable isotope labeling or by label-free quantification.
The first group of tasks is concerned with extracting
features from the raw data that correspond to peptides in the
MS spectra and to peptide fragments in the MS/MS spectra
(
Figure 1.2
A). Depending on the specific details of the MS
technology employed, pre-processing steps may be
required, for instance subtraction of a background level, or
smoothing and filtering of the raw data
[43]
. Then, peaks
are detected, which in LC-MS constitute three-dimensional
'hills' over the mass-retention time plane. These 3D peaks
usually occur in co-eluting isotope patterns that correspond