Biology Reference
In-Depth Information
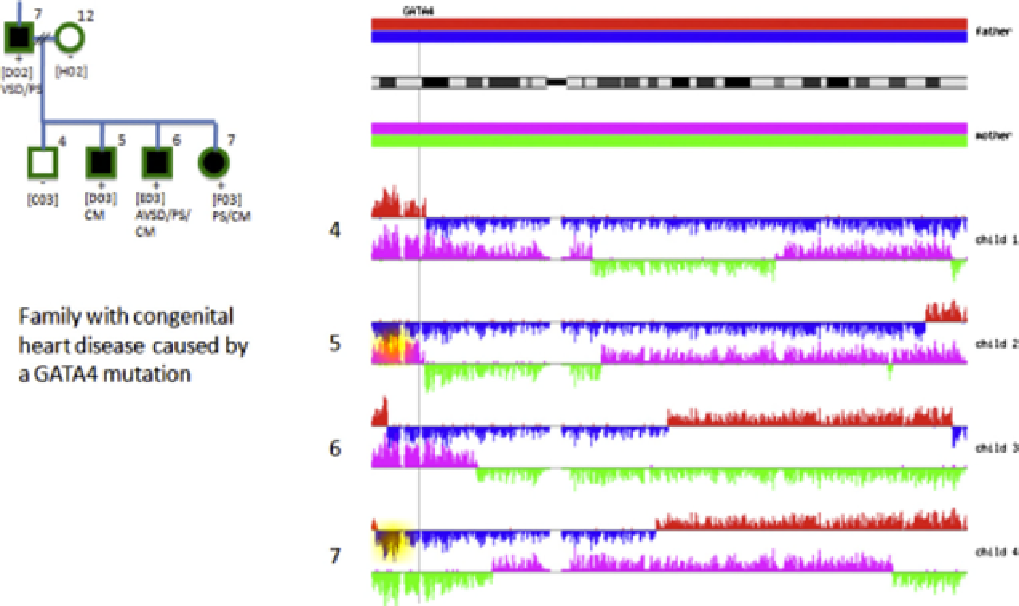
FIGURE 23.8
A schematic depicting the haplotypes of the members of a family of six. The family tree is indicated at the left. The four parental
haplotypes (two for each parent) are indicated by four different colors. The portions of the parental haplotypes that are passed on to each child are
indicated by the same colors. Each color change denotes a site of chromosomal recombination. Family genome sequencing permits one to determine these
recombinational sites with great precision. The important point is that the genes that cause particular diseases must reside in areas of shared haplotype by
those individuals in the family exhibiting the disease.
the genome, independent of any genetic models for the
two diseases. Fourth, we were able to determine the
intergenerational mutation rate for the two children (about
35 mutations per child). In this regard, it is interesting to
note that because of intergenerational mutations there is
no such thing as genetically identical twins. Finally, we
were able to reduce the candidate gene list for the two
diseases to just four possibilities. The correct defects
could readily be associated with the diseases by using
other genetic analyses of these defects. Thus whole-
family genome sequencing is a powerful approach to
enriching the signal-to-noise intrinsic to human genetic
studies. Family genome studies constitute a powerful new
approach to identifying the genome elements that are
responsible for health or disease.
The technologies of genomics are becoming increas-
ingly mature. This means that companies will be able to
perform genomic analyses far more effectively than most
academics, and these analyses will be increasingly out-
sourced to highly efficient vendors.
in several ways. First, DNA is basically digital in nature,
i.e., sequences and functions are specified by a digital
four-letter language. In contrast, proteins are synthesized
as linear structure, but fold into three-dimensional
structures to execute their functions. We can, however,
digitize the identification and quantification of proteins
through analysis by mass spectrometry (see below).
Second, proteins exhibit enormous complexity in
structures due to many different procedures associated
with their synthesis, including RNA editing, RNA
splicing, protein modification, protein processing, etc.
Indeed, some have estimated that the human genome
may produce a million or more proteins
[45]
.Third,
proteins are dynamic, changing their structures as they
execute their functions and as they interact with other
small and large molecules
[46]
. Fourth, proteins cannot
be analyzed in a global or comprehensive manner
(unlike genomic features) because of the enormous
dynamic range of protein expression (10
6
in tissues and
10
10
in blood), which exceeds the dynamic range of
detection by the mass spectrometer. Finally, there is no
protein amplification method equivalent to the poly-
merase chain reaction (PCR) of nucleic acids to enable
the amplification and analysis of rare proteins.
Mass spectrometry and the identification and quanti-
fication of proteins in proteomes. The analysis of
proteins differs from that of their genomic counterparts