Biology Reference
In-Depth Information
other proteins. For these reasons, literature-curated maps
cannot be viewed as representative samples of the under-
lying interactome, and inferring systems-level properties
from literature-curated protein
indirect, and some direct, associations that mostly do take
place in vivo.
protein interactome maps
can be misleading
[39]
. Nevertheless, literature-curated
protein
e
Large-Scale Binary Interactome Mapping
The technologies that enabled large-scale binary inter-
actome mapping were first developed in the 1990s by a few
groups
[40
protein interactome maps are instrumental in
deriving hypotheses about focused biological mechanisms.
Computational predictions have the advantage of being
applicable at genome or proteome scale for only a moderate
cost. We discuss the numerous computational strategies
that have been designed to predict protein interactions in
the section of this chapter entitled 'Drawing inferences
from interactome networks'. In brief, computational
predictions apply 'rules' learned from current knowledge to
infer new protein interactions. Albeit potent, this approach
is also intrinsically limiting since the rules governing bio-
logical systems in general, and protein interactions in
particular, remain largely undiscovered. Therefore, pre-
dicted protein
e
45]
. The following years saw significant
progress towards assembling binary protein
e
protein
interactome maps for model organisms such as the yeast
Saccharomyces cerevisiae
[39,46
e
49]
, the worm Caeno-
e
rhabditis elegans
[50
53]
, the fly Drosophila mela-
nogaster
[54]
, and most recently the plant Arabidopsis
thaliana
[55
e
57]
. Similar efforts have been deployed to
map the human binary interactome
[36,58
e
62]
.
Large-scale binary interactome mapping is amenable to
only a few existing experimental assays
[47,48,63]
and is
carried out primarily by ever- improving variants on the
yeast two-hybrid (Y2H) system
[64,65]
. The Y2H system is
based on the reconstitution of a yeast transcription factor
through the expression of two hybrid proteins, one fusing
the DNA-binding (DB) domain to a protein X (DB-X) and
the other fusing the activation domain (AD) to a protein Y
(AD-Y)
[65]
. In the last 20 years the technique has been
streamlined to increase throughput and quality controlled to
avoid foreseeable artifacts
[32,43,45,53,66
e
69]
. Today,
Y2H can interrogate hundreds of millions of protein pairs
for binary interactions, in a manner that is both highly
efficient and highly reliable.
The contemporary Y2H-based high-throughput binary
interactome mapping pipeline consists of two essential
stages: primary screening and secondary verification
[64,70,71]
. Large collections of cloned genes are trans-
ferred into DB-X and AD-Y expression vectors, then effi-
ciently screened using either a pooling or a pairwise
strategy
[49,51,64,72
e
protein interactome maps, like literature-
curated interactome maps, should be handled with caution
when modeling biological systems.
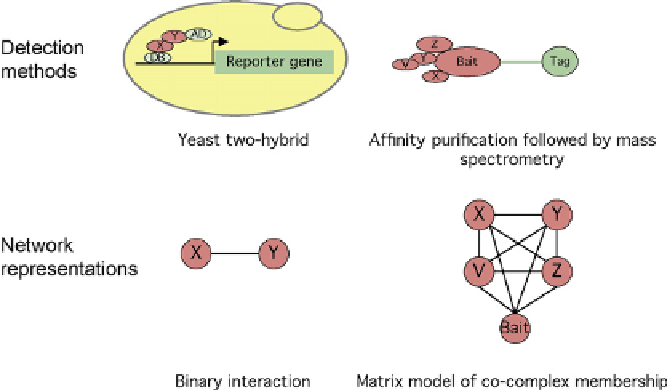
High-throughput experimental interactome mapping
approaches attempt to describe unbiased, systematic and
well-controlled biophysical interactions. Two comple-
mentary approaches are currently in widespread use for
high-throughput experimental interactome mapping
(
Figure 3.3
): i) testing all combinations of protein pairs
encoded by a given genome to find all binary protein
interactions that can take place among them and uncover
the 'binary interactome'; and ii) interrogating in vivo
protein complexes in one or several cell line(s) or tissue(s)
to expose the 'co-complex interactome'. Binary inter-
actome maps contain mostly direct physical interactions, an
unknown proportion of which may never take place in vivo
despite being biophysically true. On the other hand, co-
complex interactome datasets are composed of many
e
74]
. All protein pairs identified in
e
FIGURE 3.3
Binary and co-complex protein-
interaction detection methods and network
representations.