Biology Reference
In-Depth Information
A: Sample preparation
extraction
fractionation*
enrichment*
digestion
fractionation*
enrichment*
cells or tissue
proteins
peptides
B: Liquid chromatography - mass spectrometry
data dependent acquisiton
HPLC
ESI
top1
top2
...
top n
Full MS
MS
2
MS
2
MS
2
MS
2
Cycles of full MS followed by
MS/MS of the most intense peaks
mass spectrometer
time
C: Spectra interpretation
precursor
selection,
isolation,
fragmentation
Full MS
MS/MS
bottom-up
protein assembly
m/z
m/z
peptide m/z, peptide intensities
stable isotope-pair ratios
b- and y-ion series
reporter ions, diagnostic peaks
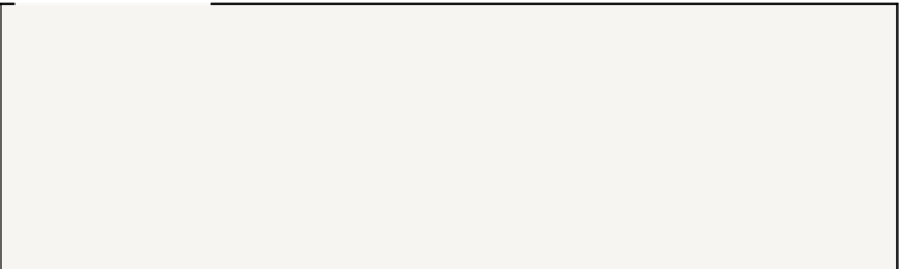
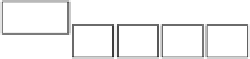
FIGURE 1.1
Outline of a typical shotgun proteomics workflow. A: Sample preparation: Proteins extracted from tissues or cells are digested into
peptides using proteases such as trypsin. A fractionation step may be applied at either the protein or peptide level to improve the coverage and dynamic
range. Peptides bearing specific post-translational modifications can be enriched using specialized approaches (see
Figure 1.5
A). B: Liquid chroma-
tography-mass spectrometry: Peptides are separated by high-performance liquid chromatography (HPLC) and electrosprayed directly into the mass
spectrometer. Peptide ions are measured at high resolution in a data-dependent mode: after each full MS scan, the most intense peptide ions are fragmented
to generate MS/MS spectra. C: Spectra interpretation: The full MS spectra provide information about the peptide mass, intensity, presence of a PTM and
stable isotope pairs. The mass of each fragmented peptide together with its fragment ion pattern is searched against databases for peptide identification and
bottom-up protein assembly.
the bottom-up principle: proteins are first digested to
peptides using a sequence-specific endoprotease. This is
typically trypsin, which cleaves C-terminal to arginine or
lysine. These peptides are analyzed by MS and afterwards
proteins are reconstructed in silico. For the general purpose
of identifying and quantifying proteins with high sensitivity
and in complex mixtures, this 'bottom-up' approach is
extremely powerful. This is due to the convenience of
handling peptides and the much superior characteristics of
the MS analysis of peptides compared to proteins. The
complementary 'top-down' approach omits the enzymatic
digestion step and analyzes intact protein species instead
[17]
. Its principal merit is that it retains information about
the entire protein (such as co-occurring modifications), but
this advantage comes at
the cost of vastly increased
experimental effort
[18]
.
For an unbiased and comprehensive analysis of the
proteome, the cell or tissue lysis method must ensure
complete solubilization of all proteins contained in the
sample. This is particularly challenging with membrane
proteins, which demand a detergent-based solubilization
method even though detergents are known to interfere with