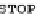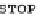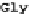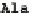Biology Reference
In-Depth Information
Sequence signals, (patterns or motifs) here are defined
as short functional DNA elements involved in the
definition of exons: the translational start site, the 5
0
or
donor splice site, the 3
0
or acceptor splice site and the
translational stop codon. Typically, these sequence
signals are represented by so-called position weight
matrices (PWMs). In these matrices, the probability of
each nucleotide at each position is computed froma set
of known functional signals. The matrices can then be
used to compute at each position along a problem
sequence the likelihood of the corresponding site to be
a functional signal. In the figure (panel 2) a PWM for
splicing donor sites is represented as a sequence logo.
In these pictorial representations, the relative
frequency of each nucleotide along each position of
the motif is represented by the height of the letter
representing the nucleotide. The total height at each
position measures the information content of the
position
models
[23]
; appear to offer the maximum discrimi-
native power, and are at the core of most popular gene
finders today. In the figure (panel 3), we have
computed a simple measure of codon bias along
a 2000 bp-long stretch of the human genome encod-
ing the
-globin 3-exons gene. We have used the
known human codon usage table (
Figure 2.2
)to
compute the likelihood that an observed sequence
occurs in a protein-coding region. We have then used
a sliding window to record the likelihood at each
position along the investigated genome sequence.
Peaks in the resulting distribution correspond to
protein-coding exons and valleys correspond to
introns.
b
Transcribed sequences. Transcribed sequences corre-
sponding to the genome being investigated are the most
powerful and reliable source of information to locate
genes in genomes (panel 4). In addition to cDNA
sequencing
l
that is, how relevant the position is in
defining the pattern.
Protein-coding regions, on the other hand, exhibit
characteristic DNA sequence composition bias,
which is absent from non-coding regions. The bias is
a consequence of the uneven usage of amino acids in
real proteins, and of the uneven usage of synonymous
codons (
Figure 2.2
). To discriminate protein-coding
from non-coding regions, a number of content
measures can be computed to detect this bias. Such
content measures
through either ESTs or RNASeq (see
previous section)
e
e
genomic similarity to known
protein-coding sequences may also provide strong
evidence of protein-coding function.
e
Integrated Computational Gene Prediction
During the last two decades a plethora of programs and
strategies have been developed to combine these sources
of information in order to obtain reliable gene predictions
(see Brent and Guigo
[24]
for a review). Programs exist
that can combine, using a variety of frameworks often
related to hidden Markov models (see Borodovsky and
McIninch
[25]
and references therein for an introduction),
the 'intrinsic' evidence from sequence signals and statis-
tical bias to produce gene predictions (panel 8). These
also known as coding statistics
e
can be defined as functions that compute a real
number related to the likelihood that a given DNA
sequence codes for a protein (or a fragment of
a protein). Hexamer frequencies, usually in the form
of codon position-dependent fifth-order Markov
e
FIGURE 2.2
The human codon usage table. For each codon (first column in each sub-table), the table lists the encoded amino acid (second column),
the relative frequency with which the amino acid is encoded by the codon (for instance, 46% of the codons encoding Phe are UUU, while 54% are UUC),
and the per-thousand usage of the codon in human coding regions (for instances, 17.6 out of every 1000 codons in human coding regions is UUU). Sub-
tables in the table are sorted by first codon position (y-axis) and second codon position (x-axis). Within each sub-table, rows are sorted by third-codon
position. Updated values taken from
http://www.kazusa.or.jp/codon/
.