Biology Reference
In-Depth Information
Synthetic Lethality Strategies for Enhancing
Anticancer Efficacy
A synthetic lethal relationship between two genes
exists when loss of function of either gene alone is viable,
butsimultaneouslossofbothcausesdeath.Synthetic
lethal approaches to cancer treatment have the potential
to deliver relatively large therapeuticwindows and signif-
icant patient benefit.
239
This approach has already shown
considerable promise in the clinic in the treatment of
BRCA1 or BRCA2 deficient breast and ovarian cancers
with PARP1 inhibitors.
240
To identify potential thera-
peutic strategies for the treatment of cancer arising from
MMR deficiency, Martin et al.
241
screened a small mole-
cule library encompassing previously utilized drugs and
drug-like molecules to identify agents selectively lethal
to cells lacking functional hMSH2. They discovered that
methotrexate, an antifolate chemotherapeutic agent,
induced oxidative DNA damage and is selectively lethal
to tumor cells with defects in the MMR gene, hMSH2.
They further exploited synthetic sickness/lethality (SSL)
as a developing therapeutic strategy for MMR-deficient
cancers and found that a deficiency inMLH1 is associated
with elevated polymerase
g
expression levels, whereas
a deficiency in MSH2 is associated with elevated poly-
merase
b
expression. Increased 8-oxoG accumulation
correlated with SSL in either MLH1- or MSH2- deficient
cells. Both SSLswere rescued by silencing the adenine gly-
cosylase MUTYH, suggesting that lethality could be
caused by the formation of lethal DNA breaks in response
to 8-oxoG accumulation mediated by MUTYH.
242
These
data suggest that synthetic lethal is a potential major
upcoming strategy for drug and siRNAdiscovery to over-
come resistance and obtain promising therapeutic effi-
cacy. Making these tumor-selective approaches will be
a major challenge, as the effects of single knockout or
knockdown genes may be toxic to specific normal cells,
such as stem cells. The high level of genetic instability
due to point mutations and small insertions/deletions
also raises potential problems for this strategy as a matter
of therapy, since the selective pressure of treatment will
“select out” resistant cell populations.
CONCLUSIONS
Recent data suggest that direct MMR-c-Abl signaling,
and not a futile cycling pathway, determines the
response of both G
2
cell cycle checkpoint arrest
responses and cell death through apoptosis after specific
alkylating agent damage (
Figure 9.4
). More specifically,
our data show that cells proficient in MMR are able to
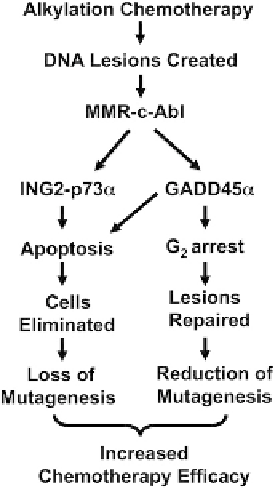
FIGURE 9.4
The MMR-c-Abl signaling cascade is not only a stra-
tegic regulatory mechanism to reduce mutagenesis in alkylated cells,
but is also a necessary signaling event required for chemotherapy
efficacy. Various DNA lesions are generated by exposure of human
cancer cells to alkylation chemotherapy (e.g., O
6
-meG after TMZ). In
cells proficient in DNA mismatch repair, the MMR-c-Abl signaling
pathway plays a key role in detecting specific lesions in order to
proofread post-replicated DNA. An under-appreciated function of the
MMR repair complex is its downstream “DNA lesion sensing/signal
transduction” system that triggers cell death in severely damaged
cells, and/or confers added time for DNA repair through prolonged
G
2
cell cycle checkpoint arrest responses. With these “additional”
functions, the MMR system efficaciously prevents mutagenesis and
emergence of highly mutagenic cells, preventing cancer initiation in
normal cells and carcinogenic progression in exposed cancer cells, in
at least three different levels: (i) the system directly repairs damaged
DNA mismatched lesions; (ii) the system initiates, maintains and
prolongs G
2
cell cycle checkpoint arrest by stimulating c-Abl-
GADD45
a
expression; and (iii) the system triggers cell death (e.g.,
apoptosis) in severely damaged cells by stimulating c-Abl-ING2-p73
a
and/or c-Abl-GADD45
a
expression. Recent data have demonstrated
that these processes, mediated by GADD45
a
or ING2/p73
a
, can be
separated using specific shRNA knockdown strategies.
90,91
These
findings strongly support the DNA damage sensing/signal trans-
duction mechanism of cell death and G
2
arrest responses directed by
MMR, and argue against a MMR-mediated futile cycle pathway
where the creation of DSBs mediates both cell death and G
2
arrest
responses and the processes would not be separable. Understanding
the link between DNA repair and downstream signaling should
elucidate drugable targets for improved cancer therapy.
Targeting “Gain-of-Function” Mutations
Since MMR-deficient cancers show “mutator pheno-
types,” a large proportion of MMR-deficient cancers
have a great chance of acquiring “gain-of-function”
mutations in oncogenes. Hence Vilar et al.
243
reported
that PI3K-AKT is a potential therapeutic target for
MMR deficient colorectal cancers. Other studies from
Solit et al.
244
reported that BRAF mutation predicts sensi-
tivity to MEK inhibition. These data highly suggested
that “gain-of-function” mutations could be another alter-
native therapeutic target for MMR-deficient cancers.