Biology Reference
In-Depth Information
physiological doses of MNNG compared to MMR-
proficient cells.
91
levels completely abrogated MMR-dependent G
2
arrest,
as well as long-term survival responses. Further studies
demonstrated that GADD45
a
-mediated MMR-depen-
dent G
2
arrest regulation after MNNG exposure, was
a downstream mediator of MMR-c-Abl signaling (see
Figure 9.1
). Importantly, experiments in these papers
also examined whether ATM/Chk2 or ATR/Chk1
played roles in MNNG-induced, MMR-dependent G
2
arrest. Even though these kinases were activated, no
specific functional roles for these activated kinases
were found. Others have found physical interactions
between c-Abl and hMLH1 and hMLH1-dependent
mitogen-activated protein kinase (MAPK)s (c-Jun,
JNK, MKK4) kinase activation after MNNG exposure.
152
Thus, based on doses of agents used and functional data
noted to date, we concluded that the MMR-c-Abl-
GADD45
a
pathway controls G
2
arrest and lethality
responses in cells under clinically relevant exposure of
DNA damaging agents.
c-Abl and MMR-dependent G
2
arrest
Data from Dr Jean Wang's lab,
101
as well as from our
group demonstrated that c-Abl was a key mediator that
regulated MMR-dependent G
2
arrest and cell death after
physiological exposures to cisplatin or MNNG.
90,91
c-Abl is a non-receptor tyrosine kinase that interacts
with a large variety of cellular proteins, including
signaling adaptors, kinases, phosphatases, cell-cycle
regulators, transcription factors and cytoskeletal
proteins.
143
e
146
It may function in a wide range of
cellular processes, including regulation of cell growth
and survival, as well as oxidative cell stress and DNA-
damage responses.
147
e
150
Although initial studies by
theWang laboratory
101,151
indicated that c-Abl was a crit-
ical mediator for MMR-dependent cellular lethality
through p73
a
after exposure to cisplatin, the role of
c-Abl was not well established. In a series of follow-up
studies, our laboratory demonstrated that inhibition of
c-Abl by the c-Abl-directed inhibitor, STI571 (Gleevec
GADD45
a
and MMR-Dependent G
2
Arrest
Although phosphorylation and stabilization of p73
a
were noted in response to MNNG exposures, a role for
this p53 family member in MMR-dependent G
2
cell
cycle arrest responses was not noted. Instead, we found
that MMR-dependent signaling led to elevated expres-
sion of GADD45
a
after FdUrd or MNNG treat-
ments.
90,91,94
Accumulated evidence suggest that the
GADD45 protein family function as cell stress sensors,
mediating their activities through a complex interplay
of physical interactions with other cellular proteins
that are implicated in cell cycle regulation and lethality
responses.
153,154
Studies using shRNA knockdown of
GADD45
a
in MMR-competent cells, as well as
complete knockout of GADD45
a
in MEF cells, further
demonstrated the role of this protein in MMR-depen-
dent G
2
cell cycle checkpoint and lethality responses
after MNNG exposures, since abrogation of GADD45
a
function resulted in loss of G
2
arrest, apoptosis and
lethality to this monofunctional alkylating agent (see
Figure 9.1
). Although downregulation of GADD45
a
in
shRNA-c-Abl knockdown clones after MNNG expo-
sure suggested that GADD45
a
was downstream of c-
Abl, direct physical interaction between c-Abl and
GADD45
a
was required to demonstrate a direct link.
A direct link between p73
a
and GADD45
a
was not
noted, suggesting two separate signaling pathways
for apoptosis and G
2
arrest responses, respectively.
Early studies from other laboratories showed that
over-expression of GADD45
a
can mediate a G
2
check-
point arrest by direct binding and inhibiting of Cdc2,
while suppression of GADD45
a
abrogated G
2
check-
pointarrestinresponsetoDNAdamageagentexpo-
sure.
137
Data from Hirose et al.,
155
suggested that p38
a
MAPK may be activated by MMR to trigger G
2
cell
),
or short hairpin RNA (shRNA) knockdown of c-Abl
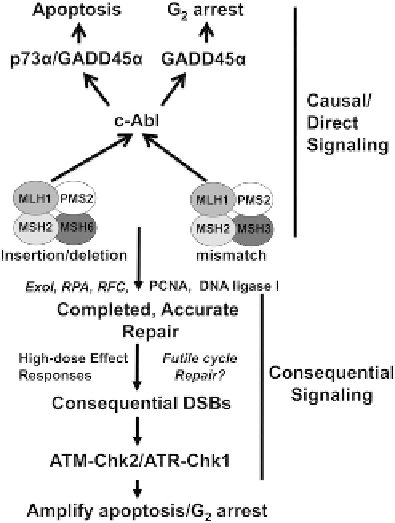
FIGURE 9.3
Signaling from MMR damage recognition and repair
to cell cycle checkpoints and apoptosis primarily involves c-Abl
activation. Recent data strongly suggest that MMR-dependent c-Abl
signaling controls cell cycle checkpoint activation at the G
2
phase of
the cell cycle, and simultaneously stimulates apoptosis in severely
damaged cells. ATM/ATR activation, on the other hand, appears to (i)
be a consequential effect of repair, possibly a result of repeated repair
cycles that are not necessarily due to MMR; and (ii) are exclusively
activated by extremely high doses of most alkylating agents, doses
that signal various repair processes beyond the low dose-sensitive
MMR activity.