what-when-how
In Depth Tutorials and Information
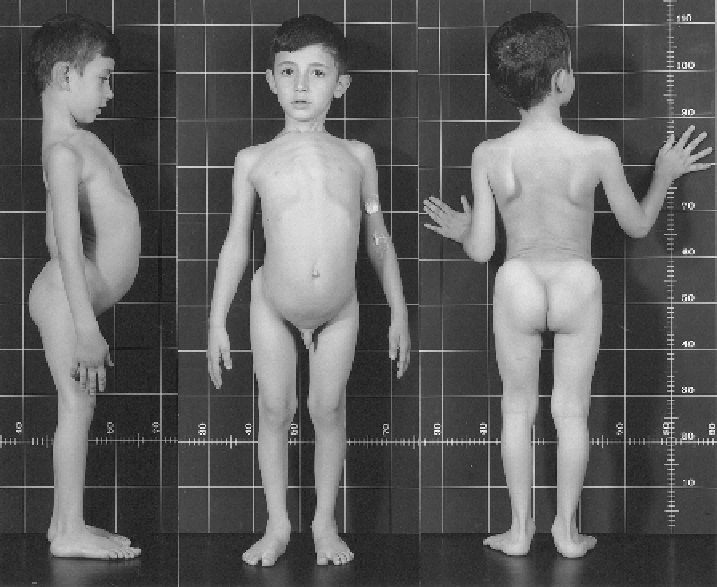
FIGURE 23.7
EDS VII-related disorder (EDS VIID). The 7-year-old patient (Case 40) presents with bilateral hip dislocation, hyperlordosis,
lat feet, pigeon breast-like deformity and scapulae alatae. His skin is moderately hyperelastic and soft, does not present abnormal scars and has
no tendency toward bruising. He has generalized joint laxity, muscular hypotonia and a waddling gait due to congenital hip dislocation. Gross
motor development was delayed. The eyes and internal organs were normal, and height and weight were on the 50th centiles. Unlike the find-
ings observed in EDS VIIA, VIIB and VIIC, collagen fibrils in the dermis were ultrastructurally normal in diameter and contour (
Figure 23.5D
).
The collagens produced by his fibroblasts were normal in amount and structure. He is therefore temporarily diagnosed as EDS VIID. Hass and
Hass
15
state that “arthrochalasis multiplex congenita is a distinct clinical entity characterized by a multiple congenital flaccidity of the joints. It
may be associated with laxity or hyperelasticity of the skin defined as Ehlers-Danlos syndrome, or it may exhibit hypermobility of the joints
without involvement of the skin.”
(Reprinted by permission from
2
.)
VIIA/VIIB but whose fibroblasts produce normal colla-
gens.
6
The normal structure of collagen I extracted from
the skin of Cases 38 and 40, produced by fibroblasts, the
absence of pNα1(I) and pNα2(I) chains in skin extracts,
normal procollagen N-proteinase activity and a normal
electron microscopic appearance of dermal collagen
fibrils (
Figure 23.5D
) clearly distinguish such patients
(Case 40 and
Figure 23.7
) from those with EDS VIIA
and VIIB. It remains uncertain whether patients such as
these are examples of EDS VII of unknown cause, tem-
porarily called EDS VIID, or whether they represent the
extreme end of severity of EDS III. It may be of note that
Cases 38-40 have smooth, hyperelastic skin but without
skin fragility.
References
Acknowledgments
We thank Wiley-Liss for copyright permission.
2
This chapter was
supported by the Swiss National Science Foundation (SNF grant
310030_138288/1 to CG).