what-when-how
In Depth Tutorials and Information
From published results
32
and the findings of Giunta
et al.,
11
it appears that open reduction with capsulor-
rhaphy, iliac osteotomy and femoral osteotomy in some
cases, is required to achieve and maintain stable reduc-
tion, and thus should diminish the frequency of hip
redislocation, recurrent dislocation, avascular necrosis
and premature osteoarthritis.
Generalized joint hypermobility is worst in infancy
when marked muscular hypotonia accompanies the
severe ligamentous laxity. Motor development is con-
sequently slow, and aids such as knee-ankle-foot and
ankle-foot orthoses are often required to stabilize the
lower limb joints for standing and walking. As muscle
tone improves, the knee-ankle-foot orthoses may be
reduced to ankle-foot orthoses.
Recurrent and/or persistent dislocations, as well as
hypermobility of upper limb joints, are also frequently
disabling.
Postural thoracolumbar kyphosis due to hypotonia
and ligamentous laxity is a feature of infants (Cases 17,
18 and 37). The spinal posture improves as the children
gain in muscle power. However, structural scoliosis has
been reported in numerous cases.
Adequate physical and occupational therapy and
orthotic management can be given to assist with stand-
ing, walking and activities of daily living.
More experience correlating detailed orthopedic
management and long-term outcome is needed before
sound recommendations can be made.
11
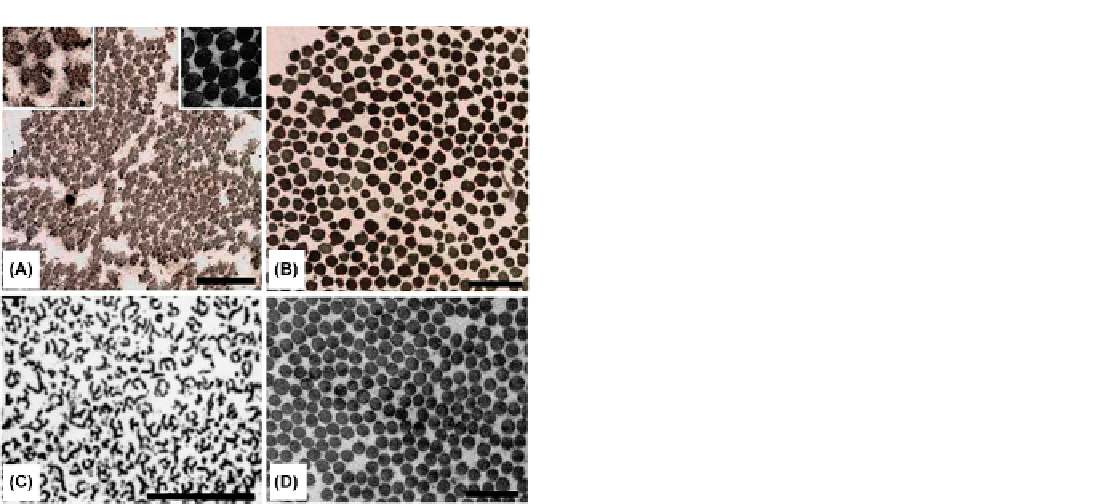
FIGURE 23.5
Ultrastructural appearance of collagen fibril cross-
sections in the dermis of a patient with EDS VIIA (A), and a patient
with EDS VIIB (B), in comparison to that of a patient with EDS VIIC (C)
and EDS VIID (D). (A) The skin collagen fibrils of an EDS VIIA patient
(Case 37) are more loosely and randomly organized, have highly
irregular contours (note the “angel-like” shape of the fibril in the left
inset) and have more variable diameters than normal (right inset). (B) A
patient with EDS VIIB (Case 7) has loosely organized fibrils with irreg-
ular contours and more variable diameters than controls; however, the
contours of the fibrils are far less abnormal than those of the EDS VIIA
patient. (C) An EDS VIIC patient presents with highly abnormal col-
lagen fibrils which have a multibranching appearance in cross-section,
resembling “hieroglyphics.” (D) An EDS VIID patient (Case 40,
Figure
23.7
) presents collagen fibrils of normal shape and diameter which are
indistinguishable from controls. Please note that the EM photographs
are at different scales (the bars correspond to 500 nm).
(A and B reprinted
by permission from
35
. C reprinted by permission from
37
.)
Future Clinical Research
We hope that in the future these rare patients
affected with the arthrochalasis type of EDS VII will
gain improved clinical awareness, especially in regards
to their bone phenotype, which should be better char-
acterized by systematic radiology, densitometry, histol-
ogy, excretion of urinary collagen crosslink products, as
well as by the rate of fracture healing.
the disorder is 50%, but prenatal diagnosis should be
possible by either DNA or protein studies on a chori-
onic villus biopsy if the defect in the index case has been
characterized. Biochemical analysis of a chorionic vil-
lus biopsy in a pregnancy at risk of the spouse of Case 7
was normal and a healthy girl was born as predicted.
38
DERMATOSPARACTIC TYPE OF
EDS (EDS VIIC)
Management
The orthopedic outcome is often unsatisfactory as
premature degenerative arthritis of the hips and other
joints occurs. The findings in 20 molecularly proven
cases of EDS VIIA and VIIB reviewed
11
show how dif-
ficult the management is and how important early diag-
nosis may be.
The principal orthopedic problem in EDS VII
patients is bilateral congenital dislocation of the hips.
To gain an insight into the management of this problem,
Giunta et al.
11
reviewed treatments and outcomes in 16
patients from 12 families.
The dermatosparactic type of EDS (EDS VIIC; MIM
225410) is an autosomal recessive trait characterized by
severe skin fragility and sagging redundant skin.
Soft, doughy skin texture, easy bruising, large umbil-
ical or inguinal hernia and premature rupture of fetal
membranes are additional clinical manifestations. It is
caused by the deficiency of procollagen N-terminal pro-
teinase (ADAMTS-2, a disintegrin and metalloprotein-
ase with thrombospondin type I motifs, MIM 604539),
the enzyme that is mainly responsible for cleavage of
the N-terminal propeptide of procollagen type I (and