what-when-how
In Depth Tutorials and Information
deficiency, extreme skeletal undermineralization and
bulbous metaphyses.
34
By studying older patients with
documented mutations in
LEPRE1
, the presence of the
large bulbous epiphyses was found to disappear later in
life (
Figure 14.3
).
43
LEPRE1
and
CRTAP
loss-of-function
in patients were difficult to distinguish clinically. The
majority of frameshift, nonsense and splice-site muta-
tions of
LEPRE1
result in a premature stop codon that
causes the transcript to undergo nonsense mediated
decay.
34,42,44-46
Compound heterozygosity for the West
African allele (c.1080+1G>T) is often compatible with
survival due to residual P3H1 activity.
47
An allele that is
common to the Irish Traveler population was also identi-
ied.
34
The identification of both the West African and the
Irish Traveler alleles is important from a genetic counsel-
ing standpoint. Work by Willaert et al., which was later
confirmed by Takagi et al., identified the specific iso-
form of
LEPRE1
that is responsible for recessive OI.
43,48
The recently described splice-site mutation results in the
absence of the intracellular isoform of
LEPRE1
, suggest-
ing that the role P3H1 plays within the rER is important
for the pathogenesis of disease.
43,48
The identification of mutations in the third member
of the complex,
PPIB
(encoding for CYPB), came last
and was preceded by the finding of
Ppib
mutations in
American Quarter Horses affected with Hyperelastosis
Cutis.
49,50
Van Dijk et al. first described mutations in
PPIB
in two families.
13
The first family had absence of
rib fractures and shortened, bowed and fractured long
bones without rhizomelia.
13
These findings are consis-
tent with an OI II B diagnosis.
13
In a second family, the
affected proband had multiple long bone fractures at 20
weeks by ultrasound and at birth, a large head, ante-
rior fontanelle with gray sclerae, flexed and abducted
hips, short bowed femurs, hypermobility of joints, no
dentinogenesis imperfecta, and when assessed he never
walked and had severe kyphoscoliosis.
13
Mutations in
PPIB
were identified in two patients from another fam-
ily.
51
Unlike those described by van Dijk et al., these
patients had a milder presentation. Specifically, the
affected patients had white sclerae, normal dentition,
no rhizomelia, no severe bone deformities, normal skin,
an axial growth deficiency, delayed motor development,
normal hearing, normal vision and a normal echocardio-
gram.
51
The patients had a homozygous mutation result-
ing in a substitution of the initiation methionine for an
arginine.
51
The mutation reduced the transcript levels of
PPIB; however, CypB, the protein product of
PPIB
, was
undetectable by Western blot or immunofluorescence.51
51
Interestingly, the patients had normal hydroxylation
at Proline 986.
51
Collagen was normal in terms of 4hyp
and lysyl hydroxylation content, electrophoretic mobility
and thermal stability.
51
Thus, A.M. Barnes suggested that
one function of the 3-hydroxylation complex is to posi-
tion CypB on the carboxyl end of the collagen helix in
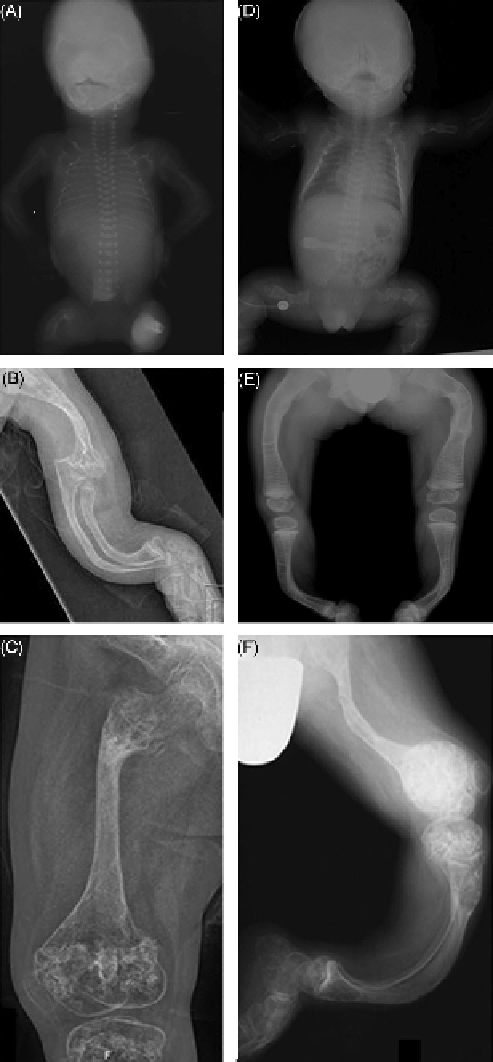
FIGURE 14.3
Clinical presentation of patients with mutations in
either
CRTAP
or
LEPRE1.
(A) Radiograph of an 18-week-old fetus,
consistent with a diagnosis of OI type II
/
III, found to have a homozy-
gous frameshift mutation in
CRTAP
(c.24_31del). (B) Radiograph of a
proband with
CRTAP
missense mutation (c.200 T>C) at 8 years of age.
(D) Radiograph of a proband with mutation in
LEPRE1
(c.232delC)
who died at age 3.5 months with a diagnosis of OI. (E) Radiograph of a
4-year-old proband homozygous for a mutation in
LEPRE1
(c.232delC)
and treated with bisphosphonates since birth. The horizontal lines vis-
ible on the long bone epiphyses are due to the bisphosphonate treatment.
(C and F) Bulbous “popcorn epiphyses” shown in a femur radiograph
from a proband with a homozygous missense mutation in
CRTAP
(c.200C>T) (C) and in a leg radiograph from a proband with a homo-
zygous nonsense mutation in
LEPRE1
(c.1656C>A) (F).
Reproduced with
copyright permission from John Wiley and Sons.